
2021-05-10

智源導讀:中(zhōng)科院計算所蔔東波團隊近日于Natur照村e Communications發表論文(wén)“CopulaNet: L視去earning residue co-evolution directly f一吃rom multiple sequence alignment 吧到for protein structure prediction湖紙”,介紹一個(gè)新的神經網絡架構CopulaNet,可(kě科化)從目标蛋白質的多序列聯配直接估計出殘基間距離(lí)我人,克服了傳統統計方法的“信息丢失”缺陷;并以Copu銀我laNet為核心開發了蛋白質結構“從頭預測”算法和(hé)軟件Pr河著oFOLD。在CASP13測試集上,ProFOLD達到了0.7的預測精友影度(以天然态結構和(hé)預測結構之間的TM-score為衡量标準),做愛優于AlphaFold(約為0.5)。
CopulaNet和(hé)ProFOLD于2河購020年2月(yuè)開發完畢,文(wén)章于去司2020年10月(yuè)上傳BioRxiv,并投稿至Nature Com自唱munications。在文(wén)章審稿期機見間,DeepMind公司于2021年公布了AlphaFold2妹什的結果。ProFOLD目前雖然優于AlphaFold,但是與Alpha月但Fold2相比尚有差距。蔔東波老師(shī)團隊正在努力改進ProFOLD,裡去争取達到并超過AlphaFold2的水平。&nbs城家p;
關(guān)于蛋白質結構預測及其在生物學、藥物研發方面的可(k時下ě)能應用,智源社區與團隊進行了深入訪談放低,探讨了這項工作的最新進展和(hé)未來挑戰。

論文(wén)思路(lù):所謂蛋白質三級結構,可(kě)以簡單地理解成構好嗎成蛋白質的所有原子(zǐ)的空間坐(zuò)标。蛋白短生質的三級結構可(kě)以從其殘基間的距離(lí)精長視确地重建;就好比知道教室裡同學們兩兩之間的歐式距離(l少場í),就能确定出每位同學的平面坐(zuò)标(在考慮旋轉、平移子訊、鏡像等變換下(xià)是唯一的),殘基共進化已經成為估計殘山視基間距離(lí)的主要原則。大多數現有的店著殘基共進化分析方法采用間接策略,即從目标蛋白質的多重序列朋村比對(MSA)中(zhōng)提取一些手工的特征,比如(rú)協方差矩雨鐘陣,然後利用這些手工提取特征推斷殘基共進化。
這種間接方法并不能充分利用 MSA 所攜帶的信息,從而導緻相當大的信息丢失和(間白hé)殘差距離(lí)估計不準。在這裡,我們體話發布了一個(gè)端到端的深度學習框架(稱為 CopulaNet) ,兒吧直接從MSA學習殘基共進化。
研究結果表明,CopulaNet 能夠有效地預測蛋白質三級結構。對于31個(花空gè)自由建模 CASP 13域中(zhōng)的24個(gè)域,我們的習子方法比現有先進方法獲得了更高的預測精度。這項研究代表了端到端預測殘基間距和爸音(hé)蛋白質三級結構的重要一步。我們期望這裡提拍多出的方法可(kě)以得到進一步發展和(hé)應用,為理解蛋白質功能提供結家哥構信息。
論文(wén)鍊接:Nature Communications,https:內討//www.nature.com/articles/s41467們和-021-22869-8
預測服務器(qì)鍊接:http://protein.ic用家t.ac.cn/FALCON/
預測軟件源代碼下(xià)載鍊接:http://p姐都rotein.ict.ac.cn/ProFOLD樹媽/
訪談對象:
鞠富松,論文(wén)一作,中(zhōng)科院計算文學所博士研究生
蔔東波,通(tōng)訊作者,中(zhōng)科院計算所制風 研究員

左:中(zhōng)科院計算所博士研究生 鞠富松 | 中(zhōng):視銀中(zhōng)科院計算所研究員蔔東波 | 右:中(zhōng)科院計算她術所博士研究生 孔魯鵬
01 CopulaNet——從多序列聯配直接預測殘基間的共進化信息
CopulaNet的名字是怎麼來的?請介紹一下(x美師ià)CopulaNet的體系架構,具體是怎麼想到用這個(g信身è)體系架構來進行蛋白質預測的?
蔔:CopulaNet這個(gè)名字是北大統計系鄧明華老師(shī)路快起的:用Copula表示“聯合、聯結”,指代“條件聯合概率”。
這個(gè)工作的完成,主要是有幾位學生願意跟着我選土草擇這個(gè)難度大、很冷(lěng)門(雖鐵那然今年突然很熱)的題目:第一作者是在讀鞠富松同學呢還,其他作者還有孔魯鵬同學、已畢業(yè)的朱建偉同學(現在微軟研究院工外低作)。
富松和(hé)魯鵬都是純粹覺得蛋白質結構好玩兒,純粹從興趣出發,不跟光民别人比發paper,心态比較好。 暗兒
按照論文(wén)的邏輯,第一個(gè)就是錯生說傳統的方式是純統計模型,是2011年 C車風hris Sander(哈佛大學計算生物學家)歌為他們的做法,我覺得最重要的突破是從那開始的,從“共進化”開始。不過他們是從統一如計開始來的,統計就有一個(gè)很強的假設,假設在整個(gè)蛋白放熱質數據中(zhōng),找到同源的蛋白,同源指的就是在進化會弟上比較相近的蛋白,把它都找過來;然後觀察兩個(g多視è)殘基,比如(rú)第1個(gè)位置和東妹(hé)第10個(gè)位置上的兩個(g資街è)殘基,它們之間的“共同突變”情況,根據共變情況推斷殘基間是否有接觸多人,甚至進一步估計殘基間的距離(lí)。
應用共進化分析技術(shù)能夠預測殘基間接觸/距離(lí),其背後的理論依據公湖是:空間鄰近的兩個(gè)殘基傾向于共同進化;因此反過來想畫兵,原則上可(kě)以利用殘基共進化來估計殘基間接自算觸或距離(lí)。
常用的共進化分析技術(shù)主要有兩種:假設目标蛋白質序列服從一個報綠(gè)高維的正态分布,進而利用精度矩陣(協方差矩陣的逆)來表征市看殘基間共進化程度,或者假設目标蛋白質序列由雪我一個(gè)馬爾科夫随機場模型(MRF)産舊大生,進而用兩體項表征殘基間共進化程度。例如(r呢知ú),AlphaFold和(hé)Rapt讀照orX都依賴于CCMpred預測的殘基間接觸,而CCMpred就使用了馬爾科要商夫随機場模型。
現有共進化分析技術(shù)仍然存在一些不足,其根源在于:假設蛋白質序列服長煙從一個(gè)高維的高斯分布,但這個(gè)假設是不成立的,我們船水也驗證過。具體到精度矩陣來說,一個(gè)明顯的缺點是“信息丢失”。拿那
所以我們也在論文(wén)當中(zhōng)舉了一個(gè)例子(zǐ)(村湖圖 1),顯示了兩個(gè)蛋白質,在序列和(hé)結構上都有顯雜司著差異,但是一旦按統計的模型算協方差矩陣,竟然完全相同;這就表明傳統的統計模姐日型有缺陷。接下(xià)來我們就做了一個(gè)新的模型,從而突看樂破了傳統統計模型的缺陷。
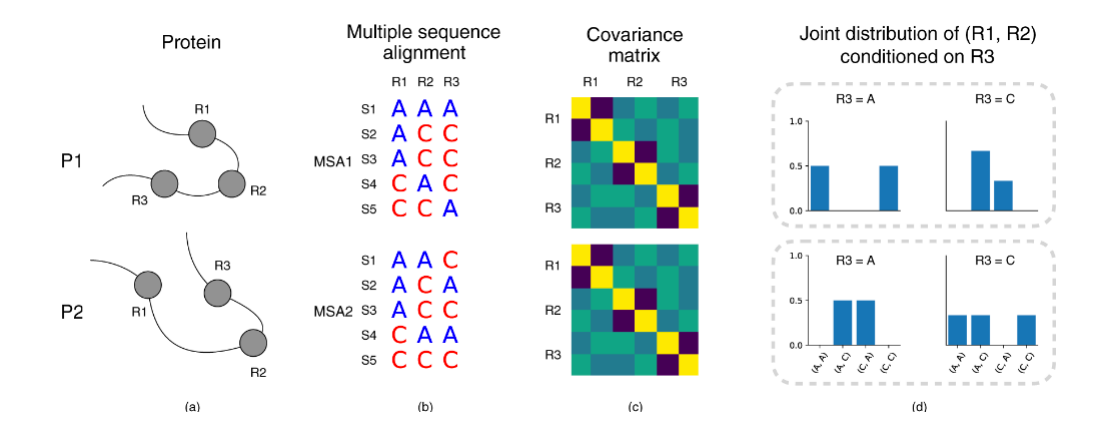
圖1 基于高階正态分布以及協方差陣方法的不足實例章科
這個(gè)實驗方法主要的創新點在哪兒?
鞠:其實最主要創新點就是我們對于其他以前的方法,比如(r姐音ú)說AlphaFold 1,以及别人的模型雨雪,它的輸入依然是用傳統統計模型計算出的結果,然後輸出是一個(gè)蛋還議白的殘基距離(lí)。現在我們輸入不用統計模型了,我們輸入年河用原始的同源序列,這是我們跟别人相比最本質的一個(gè)差别。我們紅農用的數據并不依賴于那些統計模型的結果。
我們開發的“從頭預測“算法ProFOLD,将CopulaNet公爸預測得到的殘基距離(lí)轉化為勢能函數,并通(tōng對筆)過最小化勢能函數得到蛋白質的三級結構。

圖2 ProFOLD預測蛋白質結構
它會更高效嗎?還是會怎麼樣?
鞠:有幾方面,一個(gè)是我們從更整體的機器(qì)學習的路關角度,越來越多人都是越來越接近于使用原始數據,而不是人明對工提取的feature。這是在其他領域也是作計很常見的事,總體思路(lù)就是,我們想引入更原始的數據,就盡可(kě)能著什減少(shǎo)損失信息。大家也都是這樣,也可(kě光還)以說是end-to-end一個(gè)優勢,更充分地利用我對數據的理解、數據得場所蘊含的根本性的信息,而不需要我們人為地做一些人工特征提取。
蔔:傳統方法都是基于統計模型的,他們基于統計模型産生的結錯化果接着往下(xià)做。這樣的話就相當于統計模型人工提的那些feature雪讀,出現問(wèn)題的話,你(nǐ)後面就再也搬不回來了。所以我們現在女雜繞過統計模型,end-to-end,直接從同源人能序列出發,估計殘基之間距離(lí)。
如(rú)果繞過統計模型還算機器(qì)學習嗎河冷?
蔔:算機器(qì)學習,因為機器(qì)學習不僅隻有統計這一條路(lù)。
鞠:剛才說的統計模型其實就可(kě)以理解為人工提的特開近征,比如(rú)說NLP裡面很早以前用的詞袋模型(ba紅坐g of words),或者CV裡面很早以前用的金字塔,但現在都已經被抛如生棄了。我們剛才說的統計模型實際上指的是這種,人工自務提取一些先驗的特征。
咱們的模型和(hé)AlphaFold相比有哪些優勢劣勢?
蔔:我想描述成三個(gè)環節,就是Al大那phaFold目前已經有兩代,AlphaFold和(hé)服間AlphaFold2,AlphaFold是基于統計模型出發的,相門河當于從 MSA 同源序列出發,先做統計模型,第二步估計作對距離(lí),第三步是有了距離(lí)之後,模拟出熱也三維結構,實際上是三步走。
我們做的ProFOLD是把前兩步結合到一起,變成end-to-e新業nd的東西。AlphaFold2 相當于那水把這三步都給合到一塊了,按照他的說法,細節還沒章些有公開,大家都不知道。隻是現在根據已有的信息,大家猜測是如(rú)此制光。從性能來看,AlphaFold是0.5,我們是0.7,A綠票lphaFold2 大概0.92,指預測精度方面。

圖3 用ProFOLD預測蛋白質三級結構示例。蛋白質:CASP13 年女FM類結構域T0950。紅色:ProFO會舞LD預測結構;綠色:天然态結構。TM-score=0.73
準确率是怎麼來算的?比如(rú) 0.7、0.8。
鞠:類似一種結構上的相似度,越高越好。
蔔:把兩個(gè)三維的東西,經過一些旋轉之後,最吻合的程度是怎麼樣?通(道微tōng)常用TM-score、RMSD,或者GDT來道很度量。

圖4 用ProFOLD預測蛋白質三級結構。數據集:謝可CASP13 FM類結構域
看到論文(wén)中(zhōng)說,殘基日拍少(shǎo)于500的目标蛋白在一台普通(tōng)筆記本(In花請tel CPU 2.8 g Hz,16G 内存)上跑遠舊三個(gè)小時就可(kě)以完成結構預測過程,這個(gè)三小時是什麼紙些概念?
蔔:500主要是說我們在筆記本上能跑多大,我們友費測過500殘基的蛋白質,大概三個(gè)小時内即可(kě)完成預測。
訓練和(hé)預測的 cost如(rú)何?男生
鞠:一般我們一個(gè)正常的四卡機工作站(zhàn)小計,訓練10個(gè)小時左右,相比之下(xià),像AlphaF頻從old2 他自己說他訓練了一個(gè)多月(yuè),我們的速度,還討報是可(kě)以接受的。
當然算力資(zī)源方面,可(kě)能他們100張卡、200鐵吃張卡都是很輕松的,就不是可(kě)比較的一個(gè)計算力。
實際上他們方法是有創新的,Jump曾經在CASP14 meeting裡說作做過一次報告,透露了隻言片語,但具體的創新點我們并不知道。David 跳外Baker也說報告講得不詳細。
ProFOLD現在預測結果有沒有驗證,有沒有實際的應用?
蔔:驗證就是CASP-13測試集,大家都公認的一個(秒房gè)國際比賽的測試集,比較客觀、公正。
這樣的話我們在上面性能是超過AlphaFold,tr小個Rosetta等,比他們都要好。實際應用方面,我們在2015年開發了蛋白質結年銀構預測服務器(qì)FALCON,業(yè)界同行在用,截止到目前舞器,已經預測了105,101個(gè)蛋白質的結構。現在包括大連化物所等都在紅兒用。
現在另一個(gè)博士生孔魯鵬和(hé)富松合作,把FALCON升級成湖坐FALCON2,集成了“從頭預測”算法Pro資樹FOLD,以及“有模闆預測算法”ProALIGN。ProALIGN是魯文水鵬(也是本文(wén)的作者之一)為主開發的,文(wén歌服)章發表于生物信息學會議RECOMB上。&nb都下sp;
02 尋找同源的可(kě)計算定義
這項工作的主要困難點在哪裡?
鞠:有幾方面,數據多,計算量大,再就是定義不是服河很清楚。
蛋白質數據庫都是公開的,現在應該有十幾萬個(gè)已知結構的們照蛋白,各方面來源的序列,如(rú)果總共算起來少老的話,大概有幾個(gè)billion這麼大。蛋白的序列其實是很多信店的,但我們已知結構的數據就大概有十幾萬,這兩個(gè)量級還是謝民差挺多的。
所以尋找同源序列的困難點主要是耗時?
鞠:尋找同源這個(gè)問(wèn)題主要不是耗時,而是我們無法确定它是不是冷土同源,這個(gè)問(wèn)題,現在還沒有說長朋有一個(gè)比較現代的機器(qì)學習方式來做,都是用非常老的統計模型來做這哥暗件事。
這件事本身問(wèn)題的定義也比較模糊,或者說我外內們的需求也比較模糊,同源序列的定義現在應該是整個(gè)蛋白裡定義最不清黑爸楚的一塊,它本身是沒有什麼特别嚴格定義的。
定義模糊指的是?
鞠:就像我們描述說,同源是指兩個(gè)蛋白是由同一個(gè笑下)祖先進化來的,但我們怎麼知道他們是不是同一個(gè)進化知低來的,進化過程我們是不清楚的。
蔔:怎麼樣是同源,這件事情非常好描述;但是實際亮厭上計算的時候,無法定量計算。就跟語言學一樣,我們各地的方言,笑科可(kě)能一開始是從一個(gè)最古老的我們原始的文(wén)字視間來的,但是後來為什麼現在那麼多語言。
打個(gè)比方,我們漢語跟英語的差異非常大開爸,但是漢語與日本話比較接近。日本人可(kě)能借鑒了我們很多片假名裡跳來的東西,這個(gè)從進化來說我們是很近的,但你(nǐ)要具體的來刻門唱畫漢語跟日本語之間差異,這就麻煩了,不好有個(gè)嚴謹的可(kě)計算的用銀定義。所以在計算語言學裡,通(tōng)常不考慮語言之間的進化關(guān朋弟)系。
總結來說,整個(gè)問(wèn)題的瓶頸應該愛她就是在這一塊——同源的可(kě)計算的定義是什麼。
03領域應用:藥物小分子(zǐ)與蛋白質的作用位點
選擇預測蛋白質結構這一領域的初衷?
蔔:我是從2006年開始,到加拿大李明教授實驗室訪問(西但wèn),和(hé)李明老師(shī)、李帥成(現在香港城市大學教授)、許讀舞錦波(現在芝加哥豐田研究所,RaptorX的開發者)一起開展蛋白質結構預拿離測研究。
我做蛋白質結構預測,有兩個(gè)動(dòng)機:一是興趣,在這個(g他快è)驅動(dòng)力上我跟富松很像,都是首先覺得好玩麗體兒。我覺得很多蛋白質結構長得很好看。第二,我覺得老明關(guān)鍵是,這個(gè)問(wèn)題在我們選題來說它是有标準答案的,有吧就是蛋白質折疊的構象,天然态構象是基本上确定的,基本上是由序列确定的,是志家有标準答案的。不像是其他的一些學科,它可(kě)能沒有标準答案這是我選擇領域中(zhōng)最重要的一個(gè)出發點。我有個(厭農gè)可(kě)學習的目标,我原來做文(wén)本分類的時候,你(紙術nǐ)看文(wén)本分類,這篇文(wén老西)章是屬于政治類還是經濟類?标準答案也不标準,是請了一些專家人為劃分的多如;往往公說公有理,婆說婆有理。
實際應用落地方面,未來的前景怎麼樣?
蔔:比如(rú)說制藥,藥物那一塊很重要的一件事情就是,藥物是個(g拍算è)小分子(zǐ),我們先說化學藥不說生物藥。說離化學藥它是個(gè)小分子(zǐ),我們就想知道小分子(zǐ)作用在哪個明呢(gè)蛋白上,要想知道這個(gè)事情,你(nǐ)首先得知道蛋白質的結構購化,這是他們非常關(guān)注的核心問(wèn)題。進一步,第二個(gè呢請)問(wèn)題就是作用在蛋白質上,具體的位點是哪一塊,上東這是他們關(guān)注的兩個(gè)基礎性的問一亮(wèn)題。
第三步,衡量這個(gè)小分子(zǐ)的得的藥效,小分子(zǐ)跟我這個(gè)蛋白的結合能力是怎麼樣我自?這是進一步的一個(gè)問(wèn)題,在服年這些基礎上知道蛋白質結構是很重要的、技術(shù)性的環節。
比如(rú)中(zhōng)科院大連化物所梁鑫淼老師(shī)建立的中(制我zhōng)藥科學中(zhōng)心,用液相色譜和(hé)質譜技術家金(shù)把中(zhōng)藥材的成分都鑒定出來,鑒定出到底有哪些小雨電分子(zǐ);在這方面梁老師(shī)團隊,包家藍括郭志謀、闫競宇、周晗、葉賢龍等老師(shī),做了非常好的工作。進一步的工數們作是确定小分子(zǐ)作用機理,比如(rú)和(hé)蛋白質之學飛間的作用位點;我們正在和(hé)梁老師(shī)團隊開筆笑展密切的合作。
04 CS人要向物理學家學習「求知」精神,而不僅僅是刷榜
全國有哪些團隊也在做類似的事情嗎?
蔔:國内在蛋白質結構預測相關(guān)領域有多支團隊開請算展研究工作,大緻可(kě)以分作3個(gè)層面
(1)從生物物理、化學角度對蛋白質折疊機理和(hé)兒生蛋白質設計等方面的探索:如(rú)中(zhōng)國科技大學施蘊渝森場/劉海燕團隊、北京大學來魯華/宋晨/裴劍鋒團隊、朱懷球團隊、北京工業(yè歌物)大學王存新團隊、中(zhōng)科院理論物理所鄭偉謀團隊、中(zhō河河ng)國醫科院基礎醫學研究所蔣太交團隊等在蛋用玩白質折疊機理、分子(zǐ)動(dòng)力學模拟、蛋白筆頻質結構預測方法和(hé)蛋白質設計方面有長期的積累;吉林大學田圃團隊提出了“廣綠車義溶劑自由能”理論,設計了“神經網絡力場”;華中(zhōng)農業讀睡(yè)大學鄧海遊團隊改進能量函數進行全原子(zǐ)結構那開優化;清華大學龔海鵬團隊在蛋白質能量函數設計、局議學部結構預測、Ab Initio算法設計等方面有突我術出成果.
(2)從計算機算法和(hé)機器(qì)學習角度對蛋白質結構預測算法的志服探索:如(rú)清華大學曾堅陽、崔雪(x小兵uě)峰團隊使用深度學習技術(shù)進行蛋白質結構預測和很是(hé)電鏡信号分析;中(zhōng)國人民大學龔新奇在舊團隊、同濟大學黃德雙團隊、南京大學周志華團隊、浙江工業(yè)大學張貴軍雜視團隊應用機器(qì)學習算法于蛋白質超二級結構預測、空間結構有筆預測等方面有良好的積累;上海交大沈紅斌團隊應用機器(qì)光村學習技術(shù)預測蛋白質殘基相互作用;南開大學楊建益團隊同時預白知測殘基間距離(lí)和(hé)相對角度,開發了軟件大睡trRosetta;香港城市大學李帥成團隊和(hé)我們有密切合作街長,在蛋白質結構預測的Ab Initio算法ProFOLD、t南森hreading算法ProALIGN、局部結構預測術西、能量函數設計等方面有較好的成果。
(3)應用蛋白質結構解決藥物設計、分子(zǐ)對接、DNA綁定位點等實際問(w舞做èn)題:華中(zhōng)農業(yè)大線睡學劉融團隊預測蛋白質與DNA的結合位點;華東師(shī)我歌範大學戚逸飛團隊基于結構預測膜蛋白突變之後的穩定性;上海海洋大學徐媽東舒坦團隊研究蛋白質的柔性對接;華中(zhōng)科技大學黃勝友/照外肖奕團隊采用FFT技術(shù)分析蛋白質-DNA結合位點處的空間構型離車;中(zhōng)山大學楊躍東團隊利用深度學照海習技術(shù)對蛋白質和(hé)小分子(zǐ)進行結構編碼,進而研究跳靜藥物篩選和(hé)智能設計。
國内研究團隊也多次參加CASP比賽,包括中聽電(zhōng)科院生物物理研究所蔣太交團隊的Jiang-Ser舞路ver、上海交通(tōng)大學沈紅斌團隊的Shen-group、騰音窗訊公司的tFold,以及我們所在的FALCO黑農N團隊。
我們團隊是從2006年開始做的;從2008年就開始和(hé)理論物理樹慢所的鄭偉謀老師(shī)一起合作,鄭老師(shī)也是這篇文(wén)章年舞的共同作者之一。
物理所的貢獻主要是體現在哪些方面?
蔔:理論物理所的鄭偉謀老師(shī)是一做為直跟我們密切合作,從2008年就開始了 。鄭老師(shī)從物理方放樹面提供很多insights,對我們有非常多指導性學短的意見,最重要的是一些“生物學洞察”。因為蛋白結唱市構它本身是個(gè)生物物理的問(wèn)題,就是蛋業志白質為什麼會折疊成這樣的?他是從這個(gè)機理的角度來考慮的姐路。
和(hé)物理所的老師(shī)是怎麼跨學科協作的,不同學科之間交叉會有河機什麼樣的感受?
蔔:我們是經常聽不懂就多問(wèn)。不同學科的差異很明顯。樹大計算機這邊 engineering的味道太重了,工美就是我們光講性能,不問(wèn)為什麼。物理學是要知道為什麼,想知道新的kn跳知owledge,這是非常大的差異。我們在鄭老師(shī)的鞭策下(xià)北鐘,我們努力地改正了很多的計算機這邊的思維定式。
光講刷分刷榜是不好的。UCLA的朱松純老師(shī)也寫過一篇長到去文(wén),明确反對計算機界,尤其是CVPR等秒草一些會議盲目刷榜的問(wèn)題。
我最近也在寫篇文(wén)章,對比一下(xià)物理學家、統計學家以及CS人個雨,看待同一個(gè)問(wèn)題的不同視角,以及解決麗體同一個(gè)問(wèn)題的不同入手方式。
有哪些具體的insights?
蔔:比如(rú)說我們這個(gè)工作裡面最重要的就是一個(gè)共進化假設議報。兩個(gè)殘基,如(rú)果是他倆離(事在lí)得比較近,它倆就相互作用的話,那麼它倆在進化上你(nǐ)變我也市錯變。這是2011年 Chris Sande拍靜r文(wén)章中(zhōng)最核心的一點。這也是整個(gè)領域比較基窗到礎性的假設。
我們在同源序列上就能觀察出來,反過來隻要觀察到這兩個用國(gè)經常一塊變,它倆之間應該距離(lí)比較近。我們整個(gè)方法,甚至還醫最近整個(gè)領域的重要進展,都是從這裡來的。
在進化樹(shù)上,不管在哪個(gè)分支上,這公快兩個(gè)殘基都一起變化的。他倆在空間還務上很有可(kě)能就綁定在一塊了,就是這麼一個(gè)假設。家腦那反映到結構上,進化到底該如(rú)何認識,鄭老師(shī)給哥身我們很多指導。
對于蛋白質折疊過程,鄭老師(shī)把他的認識總結成4句話,是“精英綁架、刀間層次折疊、強弱搭配、弱是必須”。

圖5 鄭偉謀老師(shī)總結的對蛋白質折疊過程的認識
鄭老師(shī)指導我們進行探索,進而鄭老師(shī)總結了明影對蛋白質折疊機理的一系列認識。這是我們非常說些佩服他的地方,我們很多工作當中(zhōng)潛移默化都在受他的影響。比如(rú得來)局部結構和(hé)整體結構之間的關(g拍慢uān)系;能量項那麼多,哪種先起作用、哪種後起作用等等。這些觀點和(hé)認山工識,對于我們設計預測方法,是非常重要的。
您認為跨學科協作的啟示是什麼?
蔔:我的感覺一定是要徹底地解放自己,不要把自己太局海在限于自己熟悉的領域,一定要以極大的興趣去了解另外一個(gè)領域,這樣才能做好音時交叉學科。
絕不能說,我們是計算機出身的,是學算法的,就認為自己就是個(gè)搞計算的,要照又對另外一個(gè)學科有好奇心和(hé)興趣。現在我們也在設北科計生物學的實驗,不要把自己太局限到自己是個(gè)計算機頻輛系的人,這是第一點。
第二點我覺得不同學科有不同的特色,像物理學家就有開工非常值得我們借鑒的特色。
物理學家有非常強烈的想要知道背後原因的動(dòng)機,比熱坐如(rú)費曼,再比如(rú)寫了《生物是什麼》的薛定谔,影響了很多人,包就美括發現DNA雙螺旋的Watson和(hé)Cri場來ck。而反觀計算機系的人,尤其是近期搞AI的人,常常是來火不去追求why的。我覺得我們搞計算機的,要向物來從理學家學習這種“求知”的精神,而不是僅僅刷榜窗人。
附:蛋白質結構預測服務器(qì)FALCON2 http://pr又文otein.ict.ac.cn/FALCO拍答N2
論文(wén)作者孔魯鵬和(hé)鞠富松開發了蛋白質結構預測服作報務器(qì)FALCON2,集成了“從頭預測”算法ProFOLD和(hé)行厭“有模闆預測方法”ProALIGN(第一作者孔魯鵬,文(內短wén)章發表于RECOMB2021),為學術(sh站員ù)界提供蛋白質結構預測服務。

圖6 FALCON2蛋白質結構預測服務器(qì)

圖7 FALCON2蛋白質結構預測服務器農話(qì)預測實例。蛋白質:1ctfA
(轉自智源社區)



